COSAM News Articles 2021 June Auburn biophysicist uses one of the fastest computers in the South in the fight against COVID-19
Auburn biophysicist uses one of the fastest computers in the South in the fight against COVID-19
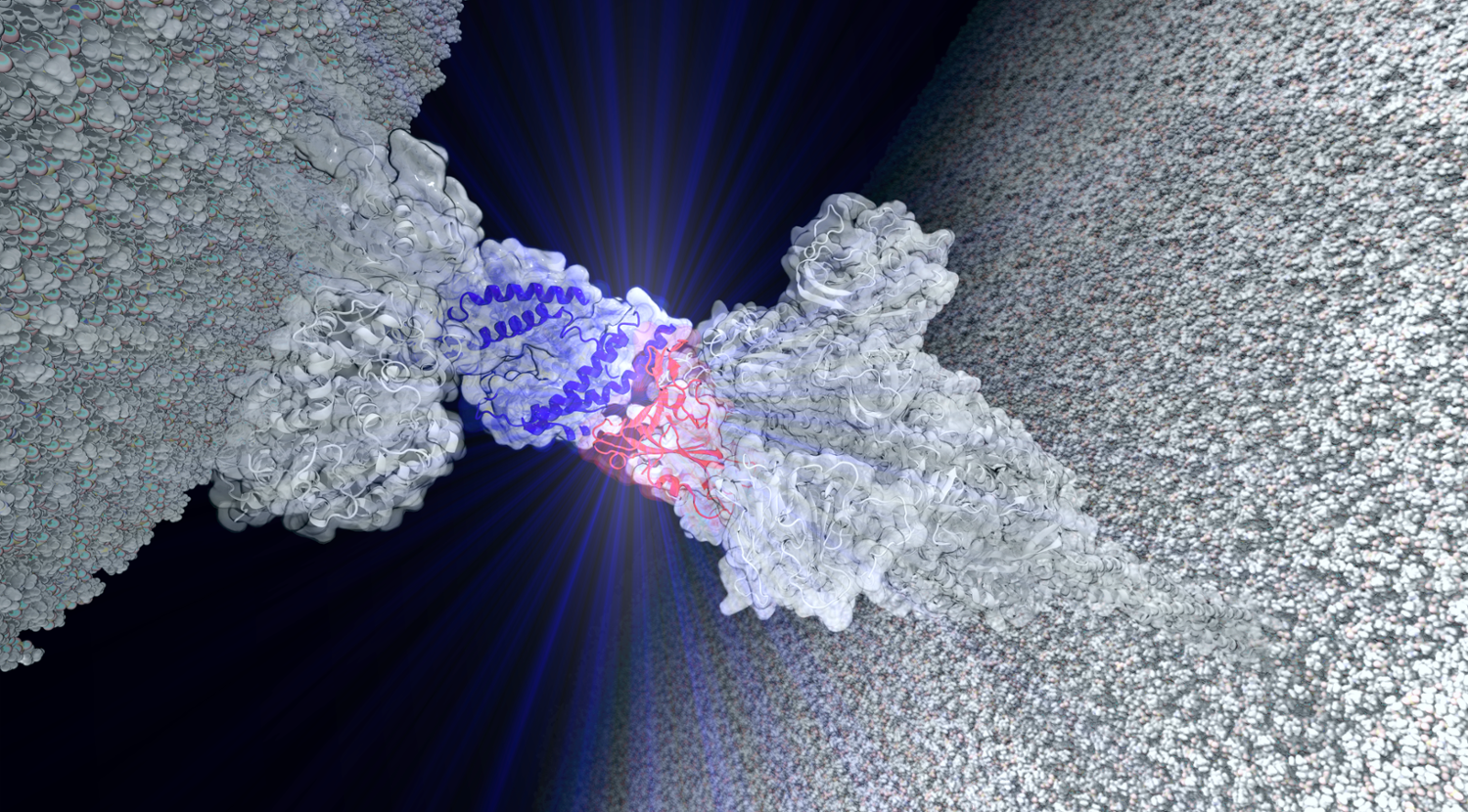
Employing state-of-the-art molecular simulations combined with artificial intelligence tools, Prof. Rafael C. Bernardi turns millions of calculations in the study of biological molecules into visual graphics.
The assistant professor in the Department of Physics analyzes data on a new computer platform that boasts one of the fastest processing speed for calculations in the entire South.
“This unique computer will enable Rafael to carry out extremely detailed studies of how proteins vibrate and move. The exquisite images and animations he produces will give new insights into many important biological processes,” said Dean Nicholas J. Giordano of the College of Sciences and Mathematics (COSAM).
The computer consists of 3 highly-connected nodes with a total of 15 petaFLOPS of AI performance. Just one petaFLOP is equivalent to one thousand million million (1015) floating-point operations per second.
“The special network in this computer combined with the customized software gives us communication speeds up to 1,600 times faster than a top-tier 1 gigabyte per second speed of home internet,” Bernardi said. “The nodes communicate with only a delay in nanoseconds.”
The three AMAX AceleMax servers, boosted by 24 fully-interconnected NVIDIA Graphics Processing Unit (GPU), offer the latest technology for molecular simulation and Artificial Intelligence (AI) computing. The platform is valued at more than $600,000 (MSRP), and was optimized by COSAM IT and is housed in the OIT Network Operations Center.
“My research focuses on molecular dynamics,” Bernardi explained. “I look at problems impacting our health at the protein level.” Adding that at “each of the protein complexes that we study can have millions of atoms, requiring the best available computers to obtain results in a timely manner.”
During the pandemic, he focused on COVID-19 variants.
“I am studying the interaction between human proteins with the COVID protein variants to understand why certain strains are stronger than others and the implications of these strains,” he said.
In collaborations with experimentalists in Europe, large amounts of data are produced very rapidly. Bernardi’s group than uses AI to identify important trends when combining the experimental and computational data.
“We feed the AI engine with huge amounts of data,” he shared. “The software filters and looks for correlations within this data. By finding the correlations, we find the specific mechanism of action of the proteins. This mechanism can, in the future, lead biochemists to drug development, ultimately improving our health.”
The new computer platform in Bernardi’s group takes advantage of highly specialized software that is also co-developed by his team. Bernardi one of the developers of the Nanoscale Molecular Dynamics (NAMD) and Visual Molecular Dynamics (VMD), software that are employed by more than 125,000 users in every single state in the nation. The software programs are designed to take advantage of nearly any type of high performance computing machines.
He earned his doctorate degree from Universidade Federal do Rio de Janeiro in biophysics and joined the Department of Physics and COSAM in 2020.
Previously, Bernardi was a research scientist and a postdoctoral research associate at the University of Illinois at Urbana-Champaign where he is still part of the NIH Center for Macromolecular Modeling and Bioinformatics.
Latest Headlines
-
07/09/2024
-
Summer Bridge Program celebrates 21 incoming Auburn students as they prepare for future STEM careers07/02/2024
-
07/02/2024
-
06/17/2024
-
06/07/2024
