


Computational/Theoretical
Department of Chemistry and Biochemistry

Associate Professor
som@auburn.edu
PhD University of Wisconsin-Madison (2006)
Disciplines: biochemistry, bioinorganic, bioorganic, biophysical, catalysis, computational/theoretical, energy, environmental, medicinal
Our laboratory utilizes a combination of bioinformatic, biochemical, and biophysical approaches to identify and characterize novel biosynthetic pathways, secondary metabolites, and biocatalysts. The systems under study are chosen for both their biological importance and their potential for employing unusual enzyme chemistry. Current projects are focused on tetrapyrroles (the pigments of life), and their applications in medical, environmental, and energy research. One example, depicted here, involves mechanistic and biosynthetic studies of dinoflagellate bioluminescence and may lead to the development of cellular imaging agents and algicides for the remediation of coastal seawaters.
James E. Land Associate Professor
emiliord@auburn.edu
PhD National and Kapodistrian University of Athens (2010)
Disciplines: physical, computational/theoretical, energy
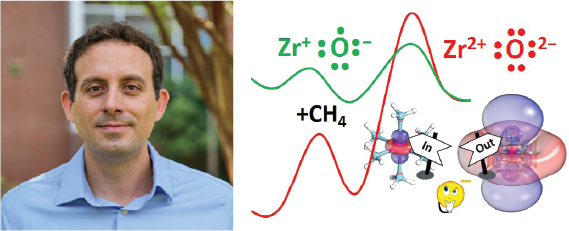
Our group applies high-level electronic structure calculations to study small transition metal compounds and their reactions with representative molecules for the activation of chemical bonds, such as C-H, O-H, N2, and CO2. These reactions are important in environmental chemistry and industry. Our goal is to understand the role of metal identity and ligands on the activity of molecular catalysts. We also study molecular systems containing solvated electrons (solvated electron precursors), which can lead to the discovery of novel materials (liquid metals) and aid their experimental characterization.

Professor and Graduate Program Officer (GPO)
patkowski@auburn.edu
PhD University of Warsaw (2004)
Disciplines: physical, computational/theoretical, energy
Our group studies weak intermolecular interactions using accurate techniques of ab initio computational chemistry. We strive to provide improved descriptions of weakly bound complexes of spectroscopic and astrophysical relevance and a quantitative picture of small molecule adsorption on carbon nanotubes and within metal organic frameworks. Our method development work focuses on extending the capabilities of symmetry-adapted perturbation theory (SAPT) and improving the performance of dispersion-corrected density functional theory (DFT) across the entire potential energy surface.

Assistant Professor
flp0008@auburn.edu
PhD Aarhus University (2004)
Disciplines: physical, computational/theoretical
Development of novel models to address a hitherto unresolved problem in quantum chemistry: Accurate and efficient prediction of a response of complex chemical systems to an electro-magnetic field. These methods can be applied to calculate virtually any property of a chemical system to very high accuracy. Examples of applications of the new methods currently underway: Excited states of biologically relevant molecules; Multi-photon absorption of potential candidates for optical materials.